click to extend
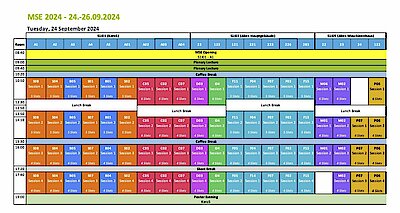
Take a look at the preliminary program of MSE 2024. All times are in CEST.
MSE 2024
24 - 26 September 2024 | Hybrid Congress in Darmstadt (Germany) & Online
MSE 2024
24 - 26 September 2024 | Hybrid Congress in Darmstadt (Germany) & Online
Subscribe to our newsletter for regular updates about materials science topics!
After subscribing, you will receive an email from us with a confirmation
link.
Only after clicking this link your registration is completed.